HLS-CATCH and TELL-Seq™: a new route to Targeted Long-Fragment Linked-Read Sequencing
At the recent 2020 AGBT meeting Universal Sequencing gave a data-filled poster on their new Transposase Enzyme Linked Long-read Sequencing kit and workflow, known as TELL-Seq™. The technology features a unique combination of bead-linked and soluble transposases that can generate linked-read libraries in 3 hours from as little as 0.1ng of genomic DNA input. Their kits offer all of the advantages of linked-read sequencing without the need for microfluidic instrumentation. The scientific poster can be viewed here.
One of the featured projects was a collaboration with Sage Science combining Sage’s HLS-CATCH targeted long-fragment sample prep with TELL-seq. Sage prepared a 200kb genomic DNA target from the BRCA1 locus (from an anonymous blood donor) on its SageHLS platform and sent the product to Universal Sequencing labs for TELL-seq sequencing. The data clearly revealed both haplotypes over the entire 200kb target region. In addition, de novo assembly of the BRCA1 reads revealed a small 1.5kb heterozygous deletion in the 5’ regulatory region of the gene.
The data demonstrated that the combination of TELL-seq and HLS-CATCH will be a powerful, high-resolution, and cost-effective option for long-range genome phasing and targeted de novo sequencing and assembly.
An example of a phased assembly from the HLS-CATCH/TELL-Seq workflow (Tom Chen, et al. presented at AGBT 2020)

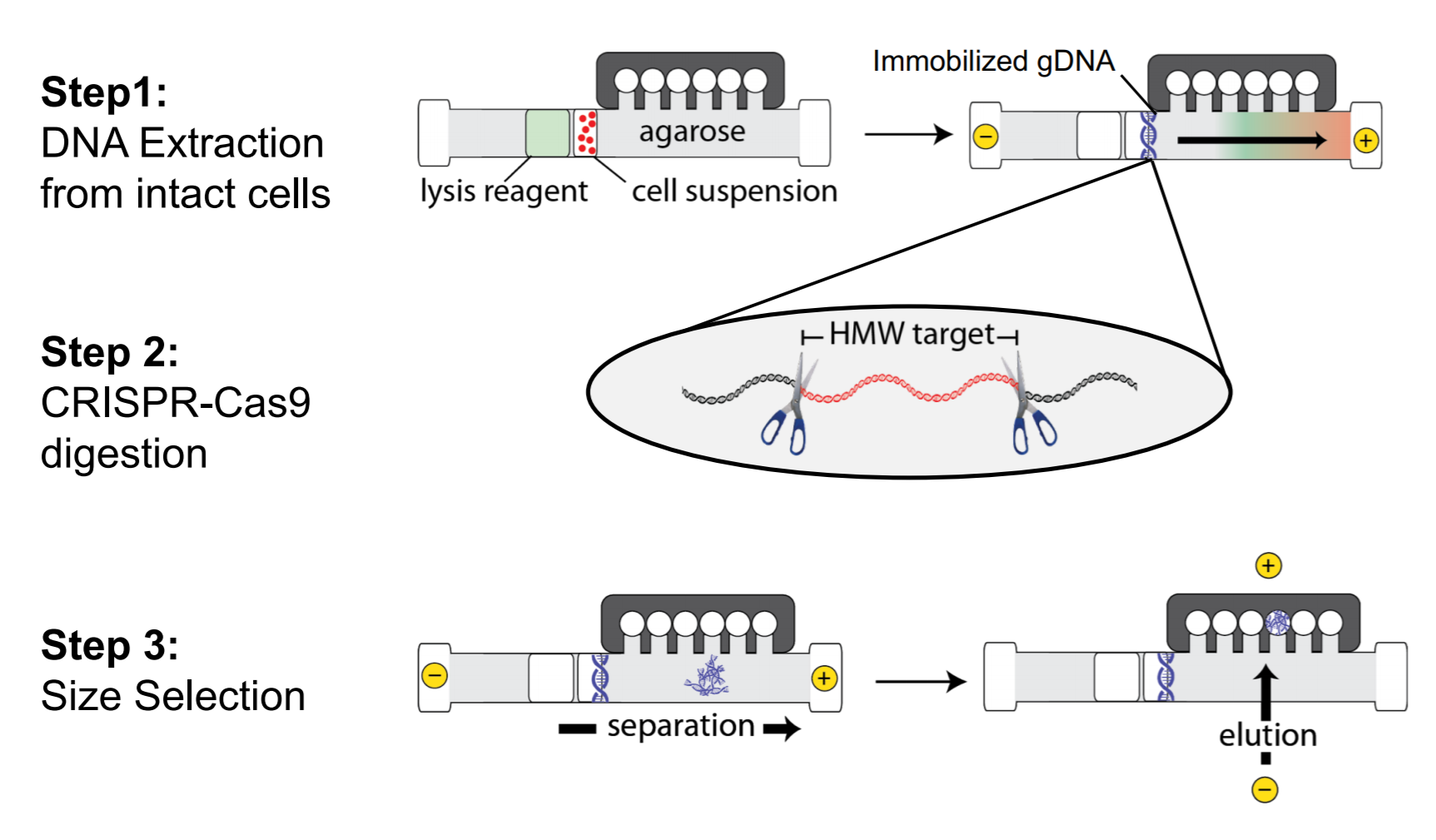
SageHLS Helps Unravel the Neuropsychiatric 22q11.2 Deletion Region1q11
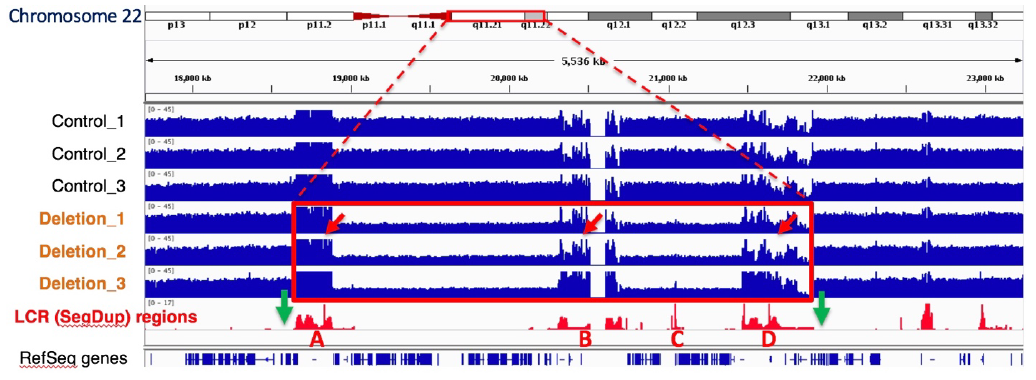
An international collaboration led by the Stanford labs of Alexander Urban and Hanlee Ji, used the SageHLS system to map a Mb-sized deletion in a complex region of the human genome, 22q11.2, that is associated with a variety of neurodevelopmental and neuropsychiatric disorders. Structural variation (SV) in this region is difficult to study because the breakpoints are frequently located in four large segmental duplications (up to 400kb in length) that are scattered over a 3 Mb area. Conventional short-read sequencing cannot unambiguously identify the breakpoints of these SVs because of the high homology between the segmental duplications. The researchers designed an HLS-CATCH procedure in which Cas9 was used to cleave unique sites outside of the 3 Mb region that contains the segmental duplications. In a patient sample carrying a large deletion, a ~300kb CATCH product from the deletion allele could be cleanly separated from the much larger 3 Mb CATCH product from the wild-type allele by preparative pulsed-field electrophoresis in the SageHLS. The researchers were then able to sequence the 300kb deletion product using Oxford Nanopore long-read methods and successfully map the deletion breakpoints.
{kind=link}
Some of this work was presented in a scientific poster at the recent AGBT 2020 conference at Marco Island. The poster can be viewed here.
The green arrows below indicate the Cas9/RNA target sites that flank the 22q11.2 region (Bo Zhou, et al. presented at AGBT 2020)
SageHLS sample prep for ultra-long Nanopore Sequencing at NextOmics/GrandOmics
NextOmics/GrandOmics is the largest third-generation sequencing service company in China. The NextOmics division focuses on animal, plant and bacterial genome sequencing, while GrandOmics focuses on human genome sequencing.
GrandOmics became the first certified PromethIon service provider in China in June, 2018. In April, 2019, GrandOmics and Oxford Nanopore announced a strategic collaboration to sequence 100,000 Chinese genomes on the PromethIon platform by the end of 2021.
Recently, NextOmics/GrandOmics has disclosed some examples of its PromethIon output on Twitter (see below), and shared with Sage Science an overview of their whole genome workflow.
1) Input material – human cultured cell lines, young plant leaves.
2) Extraction is carried out using a modified phenol/chloroform method.
3) For ultra-long reads, size selection is performed using the SageHLS instrument.
4) SageHLS input: not more than 10ug per lane.
5) SageHLS size selection program(options, depending on extraction quality): High-Pass 50kb, 100kb, 250kb, 300kb, 350kb, 500kb etc. (collection stage only)
6) Oxford library kit: SQK-LSK109 Ligation kit.
Typical size distributions from the PromethIon flow cell show read N50 values as high as 140kb, with an impressive 28% of the reads longer than 200kb.
[click the images to enlarge]
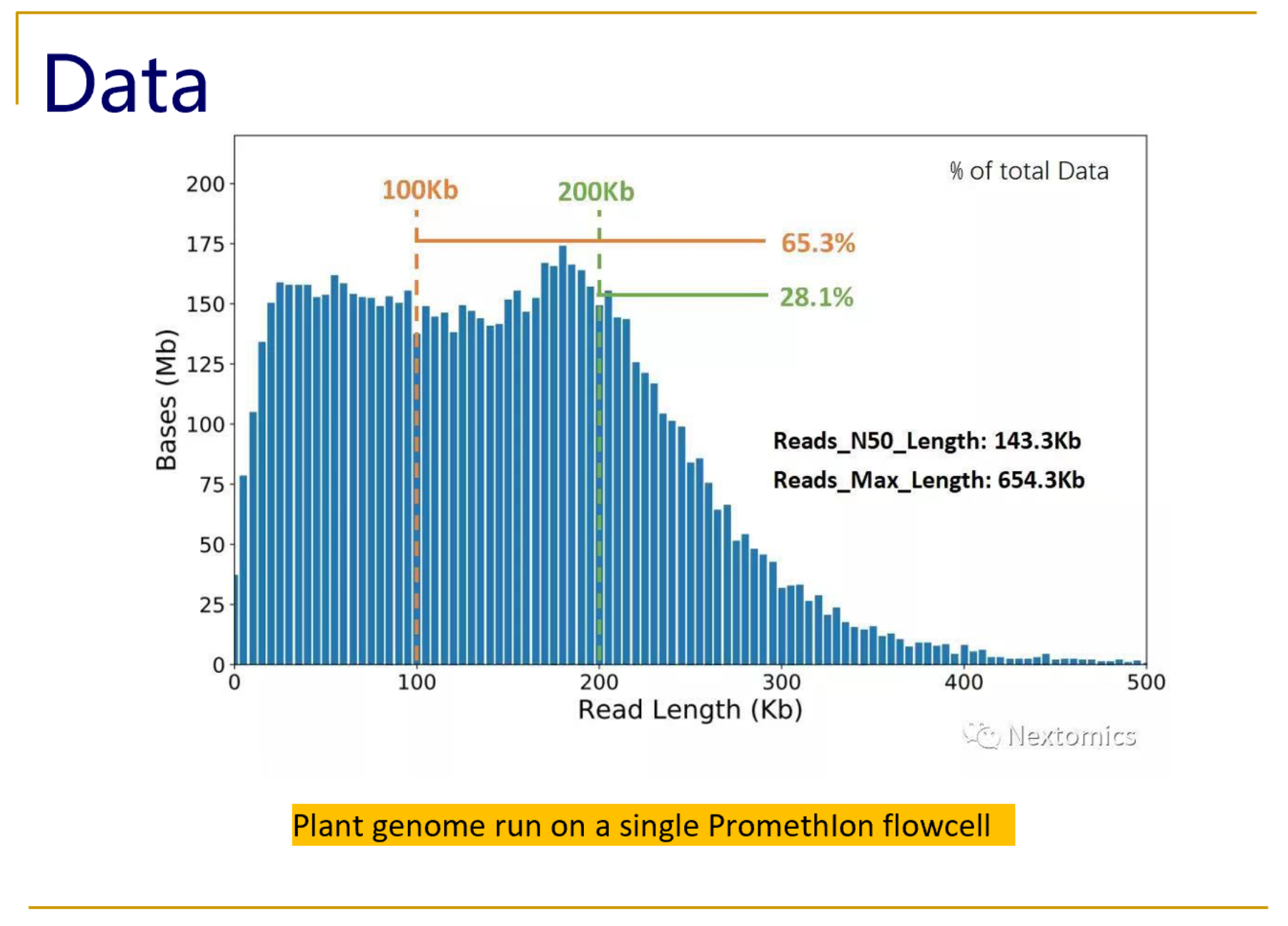
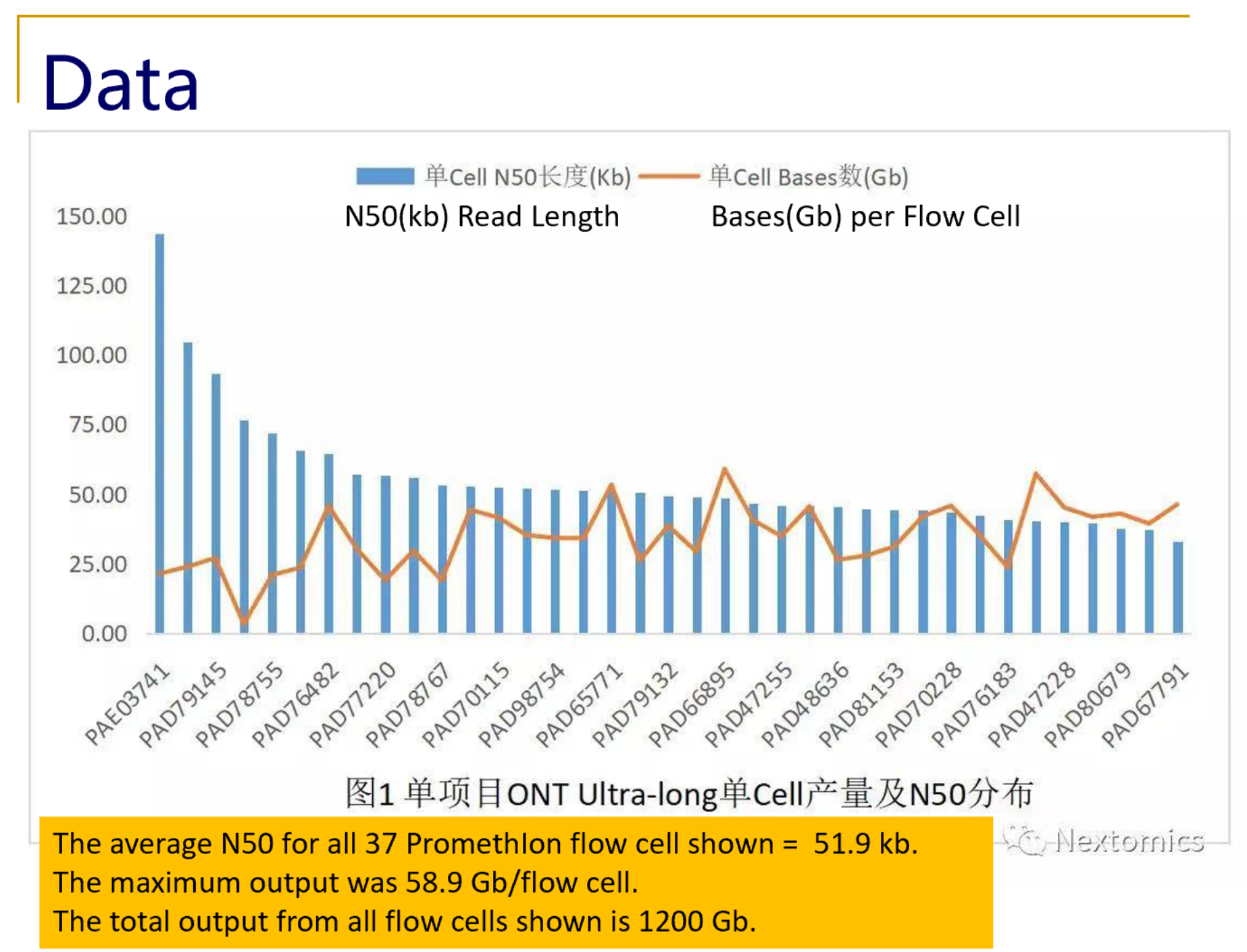
What NextOmics/GrandOmics says about SageHLS for ultra-long read PromethIon sequencing:
“The bottleneck of ONT sequencing is sample preparation, especially for ultra-long sequencing. The ONT user should optimize the extraction steps in order to protect the DNA from shear forces generated during liquid handling. After the extraction, we use SageHLS size-selection to remove shorter DNA fragments and enrich the ultra-high molecular weight DNA. SageHLS is a key component of our ultra-long PromethIon workflow.”
**We would like to express our gratitude to our great distributor, APG Bio, for their help with this post!
AGBT 2019 Re-Cap
AGBT 2019 was a smashing success this year. Featuring a triumphant return to Marco Island FL with postcard perfect weather, the science was (as usual) top-notch. The renovated Marriott greatly upgraded its conference facilities and added an attractive new tower with roof top pool and game room.
By way of recap, single-cell sequencing continues to be a dominant topic. We particularly enjoyed the talk by Jiannis Ragoussis from McGill University, providing a comprehensive expression analysis of 20,000 Glioblastoma cells, offering insight on how significant these tools can be for medical research. Spatial profiling created the biggest buzz, and the Broad Institute’s Evan Macosko’s presentation on Slide-Seq (a method where RNA is transferred from freshly frozen tissue sections onto a surface covered in DNA-barcoded beads) generated a lot of interest.
Surprising to us – if that concept can even be applied to AGBT – were the advances in genomic structure and straight up genome sequencing. We saw quite a bit of Hi-C data: Wendy Bickmore from the University of Edinburgh gave a great talk (“spatial organization of the human genome”) looking at long-range gene regulation, and Katherine Pollard from the Gladstone Institute at UCSF (“A population view of human chromatin structure”) gave a biophysical approach to examining the effect of mutations on structure. Ting Wu from Harvard provided a fascinating update (“looking at chromosomes”) on direct visualization of chromosome structure using Oligopaints and other novel methods.
For excellence in genome sequencing, NHGRI’s Adamy Phillipy’s talk, “Telomere-to-telomere assembly of a complete human X chromosome” was inspiring and used many long-range and long-read techniques, including the Oxford Nanopore Promethion (in collaboration with UC Santa Cruz’ Karen Miga, who gave a separate talk on their pipeline). Mike Hunkapiller from PacBio showed very nice gapless assemblies of the notoriously difficult SMN1 and SMN2 genes using the closed consensus sequencing method on the Sequel platform.
As for us at Sage Science, we had a relaxing beach-side Lanai suite. Aside from co-hosting a Queen-the-band themed party with Seqwell (Genomian Rhapsody), we presented a SageHLS poster that used the HLS-CATCH method to sequence the PKD1 pseudogene (our other in-suite posters can be viewed here). We believe that HLS-CATCH purification of long genomic targets can be a great tool for helping resolve difficult regions in the genome – like the aforementioned pseudogenes, other repeat elements, and SVs– and hopefully helping efforts to produce more complete genomes and understanding the function of genome structure.
Obviously, it is impossible to truly summarize such an intensive meeting- but we would like to give an additional shout out to John Charles from NASA who came down to give an update on Mars Mission plans– what could be cooler than that? On another note, next year’s meeting will mark the 20-year anniversary of AGBT. We’re sure there will be something extra-special in store, look forward to more great science!
Linked-Read Sequencing Advances Understanding of Cancers
A new study* from the University of Connecticut Medical School, Jackson Labs, and collaborators demonstrate the utility of using emulsion based linked-read sequencing (10X Genomics) for cancer research. Published in January’s Otology & Neurotology studies patients with Neurofibromatosis Type 2 (NF2) – a disease that manifests as benign brain tumors in the sheath of the cranial nerve VIII, typically causing hearing loss. The researchers compared DNA from five patients with fast-growing tumors with DNA from five patients slow-growing tumors and DNA from matching blood samples.
Using whole genome linked-read sequencing, results identified several large deletions (ranging from 5 to 650kb) in the NF2 locus correlating to the severity of the disease phenotype. The study reveals other correlating structural variants in a number of genes including FBXW7 (implicated in tumorigenesis in many other cancers) and TSPAN (implicated in esophogeal cancer). Interestingly, 4 of 5 of the high-growth tumor patients showed deletion in the VEGF-C locus. Citing a number of studies and trials of the anti-VEGF drug bevacizumab, which targets VEGF-A, the authors believe that the VEGC-C result supports previous findings that suggest that it could be predictive of treatment response.
From a methods standpoint, linked-read sequencing requires only 1 ng of DNA input and produces haplotype phasing information. For this study, our PippinHT was used to filter away smaller DNA fragments (using the >40kb High Pass protocol) to maximize the efficiency of the linked-reads for long range SV analysis. With a 1 ng input requirement, there is ample recovery in the PippinHT – users simply quantify the sample with a Qubit fluorometric assay, and dilute the sample accordingly. Concentration or buffer exchange is not necessary. On a side note, our SageHLS platform has the capability to provide very large targeted genomic regions, a great application for linked-reads given the low input requirement (read a preprint about this here).
*Linked-read Sequencing Analysis Reveals Tumor-specific Genome Variation Landscapes in Neurofibromatiosis Type 2 (NF2) Patients
Roberts, Daniel S. et al., Ontology & Neurotology: February 2019 – Volume 40 – Issue 2 – p e150-e159
doi: 10.1097/MAO.0000000000002096